5.3 实验数据的特征转换 EMP_feature_convert
在组学研究中,基因的命名方式因所使用的数据库不同而不固定。因此,在进行转录组学富集分析时,需要将基因名称转换为其对应的唯一编号(通常称ENTREZID)。而在代谢组学分析中,同样存在着多个数据库之间的编号转换问题。EMP_feature_convert提供了基因ID和代谢物ID在多个数据库之间的便捷转换。
5.3.1 用于基因ID的转换
🏷️示例:
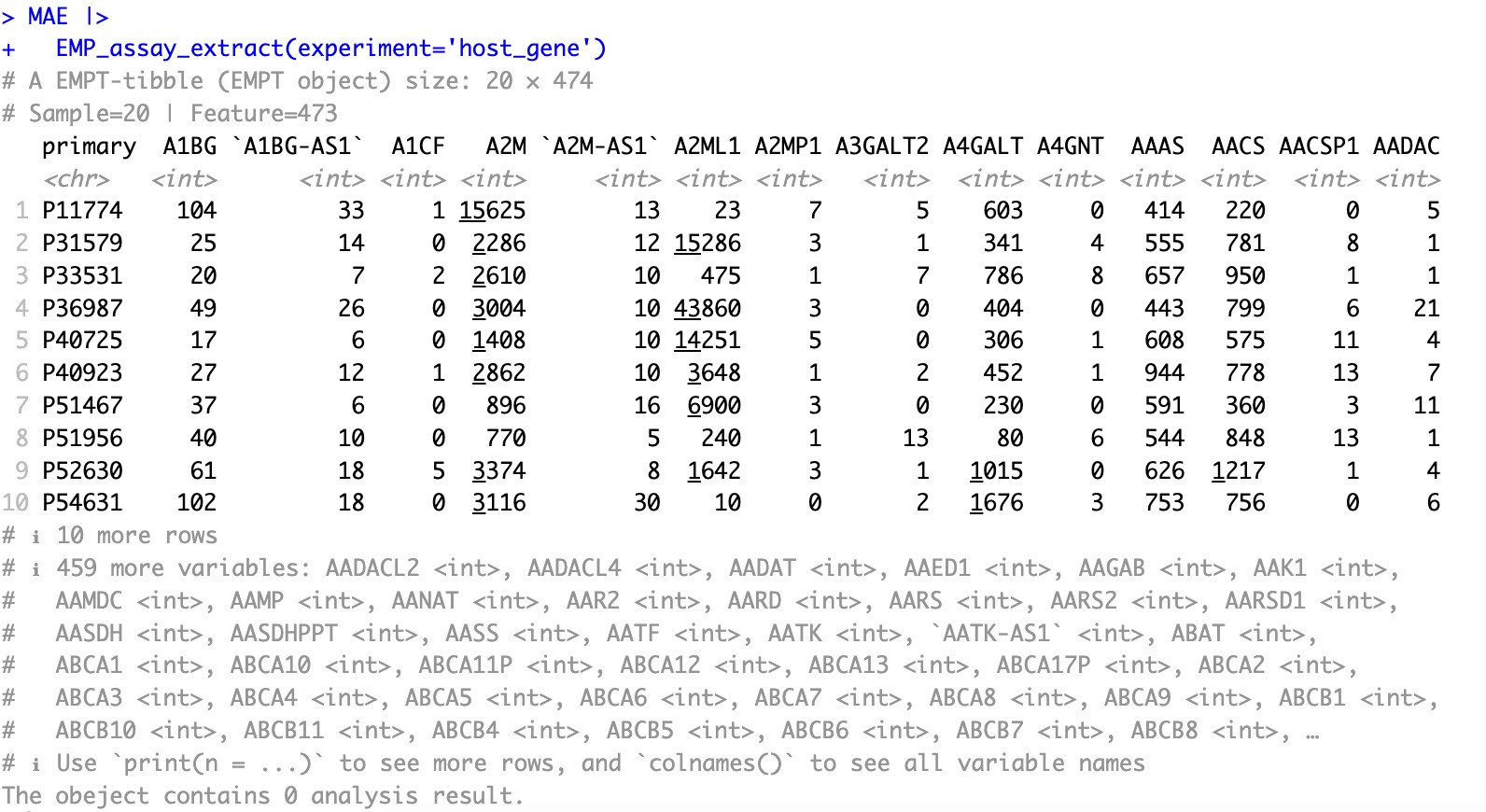
转换前:
MAE |>
EMP_assay_extract(experiment='host_gene')
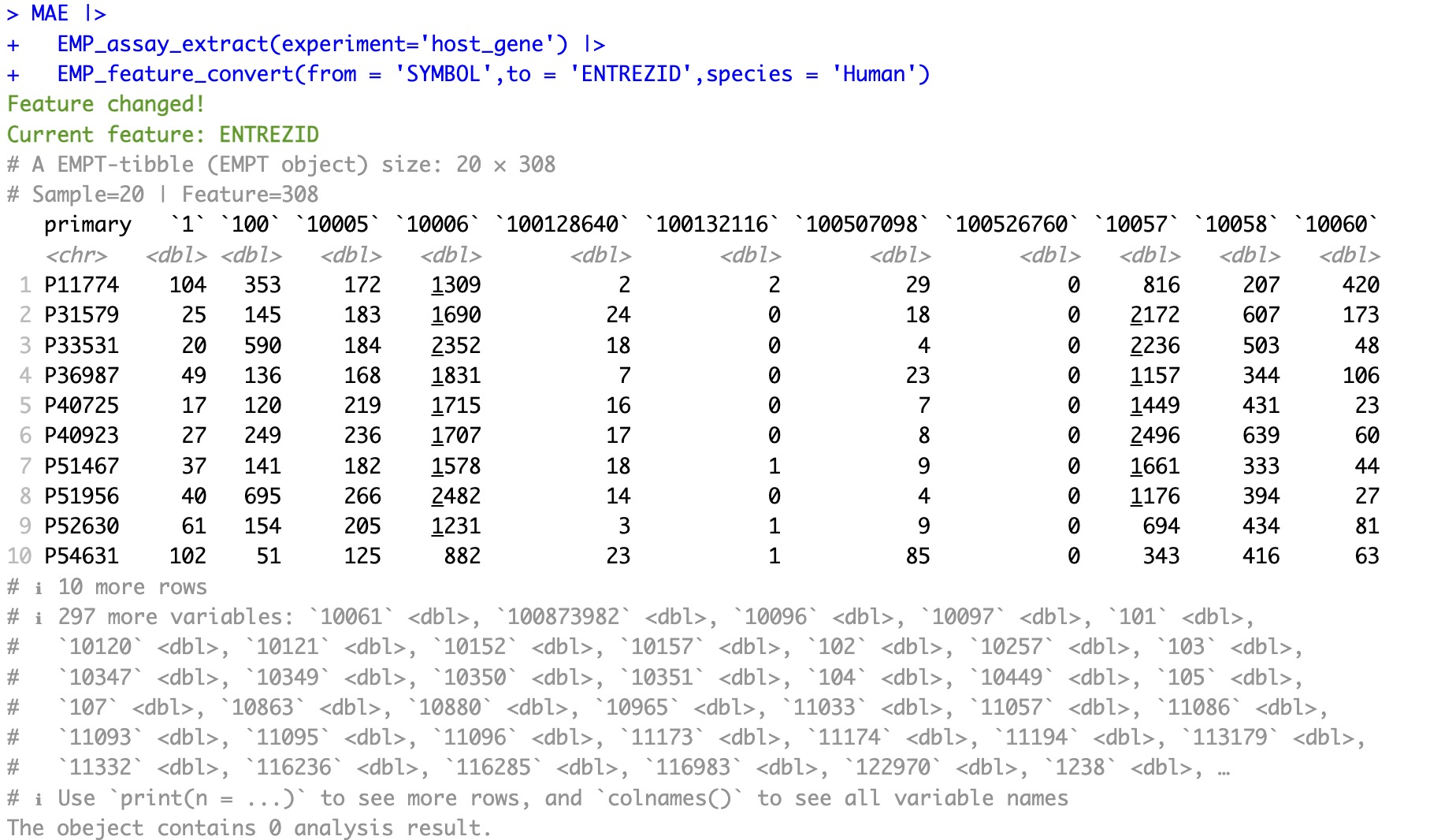
转换后:
MAE |>
EMP_assay_extract(experiment='host_gene') |>
EMP_feature_convert(from = 'SYMBOL',to = 'ENTREZID',species = 'Human')
5.3.2 用于代谢物ID的转换
🏷️示例:
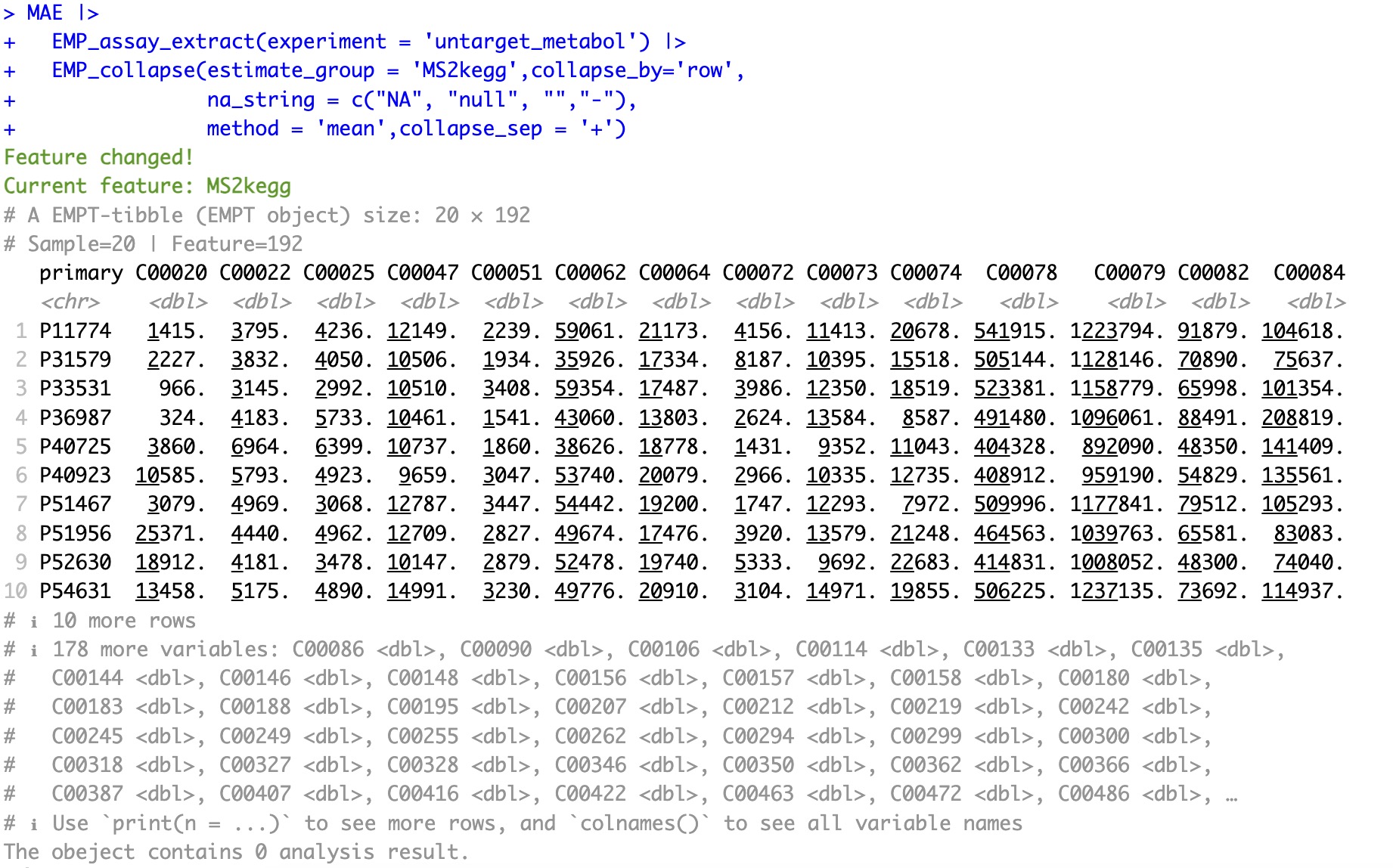
转换前:
MAE |>
EMP_assay_extract(experiment = 'untarget_metabol')|>
EMP_collapse(na_string=c('NA','null','','-'),
estimate_group = 'MS2kegg',method = 'sum',collapse_by = 'row')
转换后:
将代谢产物从KEGG的Compound注释,转换为Human Metabolome Database的ID注释。
MAE |>
EMP_assay_extract(experiment = 'untarget_metabol') |>
EMP_collapse(na_string=c('NA','null','','-'),
estimate_group = 'MS2kegg',method = 'sum',collapse_by = 'row') |>
EMP_feature_convert(from = 'KEGG',to='HMDB')

5.3.3 微生物物种注释转换
微生物一般分为界门纲目科属种等多级注释。在分析流程中。经常会将其按照不同级别进行数据折叠,进行下游数据分析。但是由于存在个别重复注释,例如某些菌的属级注释一致而科级注释不一致的情况,因此本功能可以将各个级别微生物数据补全完整注释。(详细说明在 10.4章节)
🏷️示例:
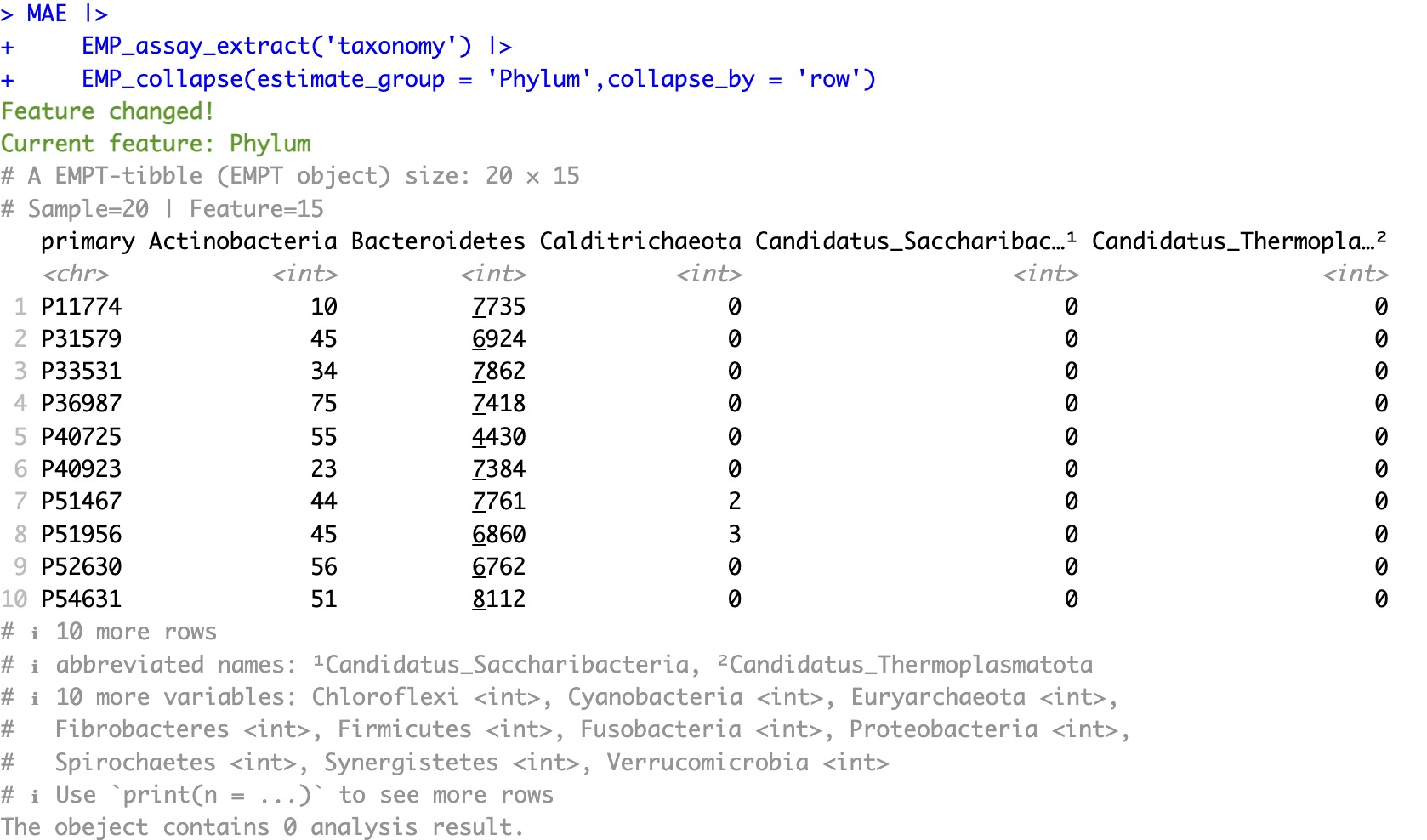
转换前:
MAE |>
EMP_assay_extract('taxonomy') |>
EMP_collapse(estimate_group = 'Phylum',collapse_by = 'row')
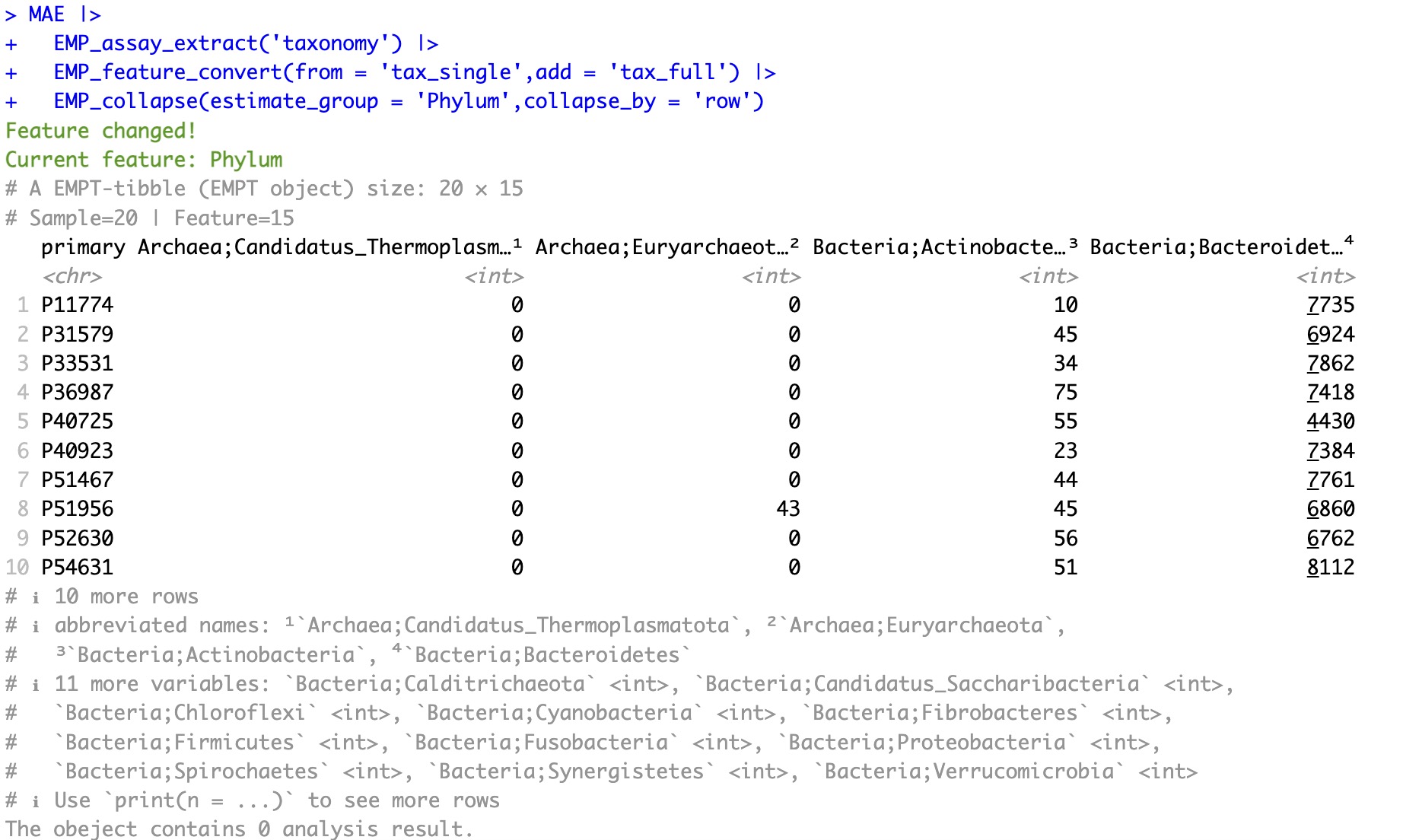
转换后:
注意:
此函数仅能在微生物数据折叠前使用。
此函数仅能在微生物数据折叠前使用。
MAE |>
EMP_assay_extract('taxonomy') |>
EMP_feature_convert(from = 'tax_single',add = 'tax_full') |>
EMP_collapse(estimate_group = 'Phylum',collapse_by = 'row')
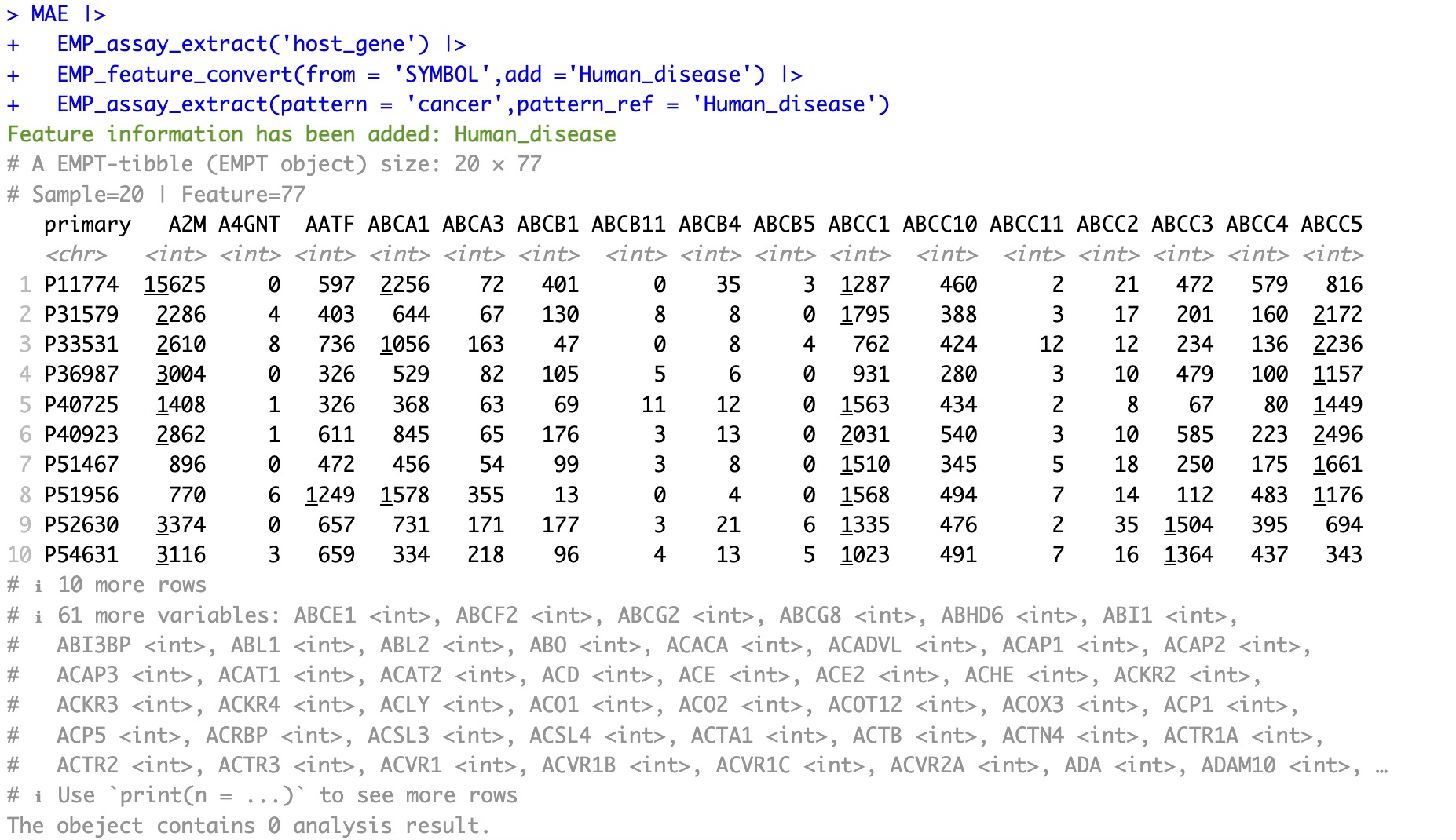
5.3.4 增加特征相关疾病注释
本模块支持对SYMBOL,ENTREZID,ko 和 ec编号增加相关疾病注释。
🏷️示例: 增加人类疾病注释,并筛选出与癌症相关的基因
MAE |>
EMP_assay_extract('host_gene') |>
EMP_feature_convert(from = 'SYMBOL',add ='Human_disease') |>
EMP_assay_extract(pattern = 'cancer',pattern_ref = 'Human_disease')